2025年11月12日,中国科学技术大学生命科学与医学部、免疫应答与免疫治疗全国重点实验室、合肥综合性国家科学中心大健康研究院曹灿教授课题组与中国科学技术大学生命科学与医学部薛天教授课题组以及天津医科大学陈贺特聘研究员课题组联合研究团队,在Cell杂志发表题为“Pathway-Selective 5-HT1AR Agonist as a Rapid Antidepressant Strategy”的研究文章。该研究提出了一种基于5-HT1A受体信号通路选择性的快速抗抑郁新策略,并据此理性设计出候选化合物TMU4142。实验结果显示,TMU4142在抑郁小鼠模型中展现出显著的快速抗抑郁样效果。该策略直击临床上选择性血清素再摄取抑制剂(SSRIs)类抗抑郁药物普遍存在的起效缓慢难题,为新一代快速抗抑郁药物的研发提供了崭新的思路和可行路径。

抑郁症已成为全球最主要的精神健康负担之一,影响超过3亿人,并呈持续上升趋势。抑郁症的发病原因复杂,目前科研界的主流观点之一是抑郁症的发生与大脑中一种名为5-羟色胺(5-HT)的神经递质水平降低有关。相应地,针对该理论开发的选择性5-羟色胺再摄取抑制剂(SSRIs)可以阻断5-HT转运蛋白对神经突触间隙5-HT的回收,进而增加突触间隙中的5-HT浓度来达到抗抑郁效果。与之前的其它抗抑郁药物相比,SSRI类抗抑郁药物相对安全,副作用往往较少。自上市以来,SSRI类药物迅速成为最为流行的抗抑郁药物。抗抑郁药物的五朵金花(氟西汀、帕罗西汀、舍曲林、西酞普兰和氟伏沙明)全都是SSRI类抗抑郁药物。但SSRI类药物通常起效较慢,需要2-4周的持续服药将中缝核突触前膜的5-HT1A受体脱敏之后,才能发挥一定的抗抑郁作用。而在这期间,由于5-HT1A自受体介导的负反馈调控,大脑中的5-HT水平会下降,抑郁症患者的症状反而可能会加重,在开始用药后几周内抑郁症患者甚至可能会出现自杀风险。这种风险在儿童、青少年和年轻成人(≤24岁)中更加明显。因此,多数主流的抗抑郁药物都有FDA的自杀风险警告。
与中缝背核的5-HT1A自受体负反馈抑制所引起的抗抑郁效果延迟相反,位于海马体和前额叶皮层的5-HT1A异源受体(5-HT1A heteroreceptor)的激活可以产生强效的抗抑郁效果。虽然是同一个蛋白,但由于5-HT1A自受体和5-HT1A异源受体分布的脑区、调控的神经环路及神经递质不同,在抑郁症的治疗中发挥着截然不同的作用。因此,如果能够设计出一个可以选择性激活5-HT1A异源受体的5-HT1AR激动剂,则有可能摆脱中缝背核5-HT1A自受体在抑郁症治疗中的负反馈作用,进而发挥出快速抗抑郁的效果。但一个关键的问题是,5-HT1A自受体和5-HT1A异源受体只是分布在不同脑区的同样蛋白,它们具有相同的蛋白序列和三维结构,理论上没有任何小分子药物可以差异性的结合这两种受体。在过去的几十年中,针对该方向的诸多努力都以失败而告终。
在过去的研究中,人们逐渐发现5-HT1A受体在不同的脑区可以偶联不同的Gi/o蛋白亚型来发挥功能。在中缝背核区域,5-HT1A自受体主要偶联Gi3蛋白。而在海马体和前额叶皮层,5-HT1A异源受体则主要偶联GoA蛋白(大脑中表达最高的G蛋白亚型)。基于这一机制,若能设计一种GoA选择性的信号偏好性激动剂,在维持对GoA蛋白激活能力的同时,弱化其对Gi3蛋白的激活,即有望实现对5-HT1A异源受体的功能选择性激活,从而提高药物的抗抑郁疗效并缩短起效时间。
此外,5-HT1A自受体所在的中缝背核区域有着大脑中浓度最高的内源性5-HT,而在海马体和前额叶皮层中,内源性的5-HT浓度是非常低的。药理学研究表明,部分激动剂在高内源性激动剂区域可表现为拮抗作用,而在低浓度区域则可表现为激动作用。因此,若能设计出效能(efficacy)较低的部分激动剂,同样有望避免激活中缝背核中的5-HT1A自受体,进而选择性地激活海马体和前额叶皮层中的5-HT1A异源受体,实现快速且显著的抗抑郁效果。
综上所述,具有GoA选择性且为部分激动剂特征的5-HT1A受体激动剂,理论上可实现5-HT1AR的脑区差异性激活,从而诱导出强效快速的抗抑郁效果。
为了筛选具有理想信号特性的激动剂,研究团队采用基于BRET2技术的G蛋白解离实验,系统性地分析了七种5-HT1A受体激动剂在不同Gi/o蛋白亚型上的信号传导谱。结果显示,包括抗抑郁药物吉哌隆(gepirone)、维拉唑酮(vilazodone)以及抗焦虑药物丁螺环酮(buspirone)在内的多种已上市药物,均能强效激活5-HT1A受体耦联的多个Gi/o蛋白亚型。具体而言,buspirone、gepirone和vilazodone等传统5-HT1AR配体在GoA亚型上表现为完全激动剂,同时对Gi3亚型也具有显著的激活作用。这种较强的Gi3活性提示它们可能高效激活中缝背核中的自受体,从而导致其在临床治疗中出现起效延迟的现象。在所有测试的化合物中,吲哚洛尔(pindolol)呈现出独特的信号特征——它在GoA亚型上仅表现为部分激动剂,而对Gi3亚型的激活作用几乎可以忽略,正符合我们所期望的5-HT1AR激动剂信号特征。值得注意的是,既往研究发现,pindolol作为SSRI类药物的辅助用药(add-on drug)可增强其抗抑郁疗效,这一现象可能与其较弱的Gi3激活能力密切相关。然而,pindolol本身是一种β-肾上腺素受体阻滞剂,对5-HT1A受体的选择性较低;同时其作为部分激动剂的效能有限,难以单药发挥理想的抗抑郁作用,因此仅在临床研究中仅被用作SSRI类药物的辅助治疗。
接下来,研究团队着手研究以下3个问题:1,为什么pindolol激活5-HT1A受体GoA信号的能力比激活Gi3信号强?2,和传统的5-HT1AR小分子激动剂相比,为什么仅有pindolol对5-HT1AR展现出较弱的激活能力?3,pindolol作为β-肾上腺素受体阻滞剂,在结合5-HT1AR的同时也对β-肾上腺素受体具有较高的亲和力,我们能否设计出对5-HT1A亲和力更高的pindolol衍生物来增加其对5-HT1AR的选择性?
为了回答第一个问题,研究团队首先解析了小分子配体结合状态下的5-HT1AR–GoA复合物以及5-HT1AR–Gi3复合物结构。结构比对结果显示,由于GoA蛋白中α4–β6环(α4–β6 loop)较为短小,5-HT1AR在结合GoA时,其跨膜螺旋TM6无需发生大幅度构象变化即可完成激活,从而使受体更易进入活化状态。除此之外,GoA的α5螺旋N端存在一个N330–H322氢键,可有效稳定α5螺旋在受体结合构象中,从而进一步促进其与5-HT1AR的结合。而在Gi3中,对应的N330位置被一个带正电的赖氨酸(K330)取代,该正电位点会与H322产生静电排斥,削弱α5螺旋的稳定性,进而降低Gi3与5-HT1AR的结合效率。将Gi3上对应的区域换成GoA的氨基酸序列,则pindolol激活Gi3突变体的能力会加强。相反地,将GoA上对应的区域换成Gi3的氨基酸序列,则pindolol激活GoA突变体的能力会减弱。令人意外的是,传统上认为负责G蛋白选择性的α5螺旋C末端对Gi/o蛋白亚型的选择性却没有显著影响。
为了回答第二个问题,研究人员解析了(S)-pindolol与5-HT1AR的复合物结构。结构分析显示,(S)-pindolol以一种独特的方式结合在5-HT1A受体中。其中,(S)-pindolol的脂肪链上的质子化N原子可以与TM7的N386形成独特的极性相互作用,而在其它5-HT1AR强效激动剂中,质子化的N原子与N386的距离相对很远,难以形成有效的相互作用。此外,不同于内源性激动剂5-HT,(S)-pindolol的吲哚环翻转180度与5-HT1AR结合,吲哚环上的N原子与S199形成了一个独特的近距离相互作用。 (S)-propranolol与S199的接触增加,则会导致ligand的efficacy下降,形成更弱的partial agonist。相反,将(S)-pindolol上的五元环去掉以减少配体与S199的接触,则会导致ligand的efficacy上升,形成更强的partial agonist。这些研究表明,(S)-pindolol与N386和S199所形成的2个独特的极性相互作用是导致其产生partial agonist活性的主要原因。
针对第三个问题,研究人员以(S)-pindolol为分子骨架,基于结构信息设计并合成了一系列(S)-pindolol衍生化合物,经过多轮的筛选和优化,最终获得了化合物TMU4142。该化合物对5-HT1A受体GoA通路的激活能力与内源性激动剂5-HT相当,而对Gi3通路的激活能力显著减弱,实现了GoA偏向性的信号转导。与(S)-pindolol相比,TMU4142对5-HT1AR的激活能力提升约15倍,而对β-肾上腺素受体的激活能力下降12–20倍。同时,TMU4142对其他12种5-羟色胺受体和5种多巴胺受体均无激动活性,展现出优异的受体选择性。值得注意的是,TMU4142在5-HT1A受体中的结合模式与以往已知的5-HT1AR激动剂显著不同。其分子头部插入了一个称为EBP3(extended binding pocket 3)的高度特异性结合口袋中。该结合口袋的关键氨基酸残基在5-HT1AR中高度保守,这也解释了TMU4142为何能够在众多五羟色胺受体中表现出极高的选择性。
鉴于TMU4142突出的药理特性,研究人员进一步评估了其在强迫游泳实验(forced swimming test, FST)和悬尾实验(tail suspension test, TST)中的抗抑郁效果。结果显示,1.5 mg/kg剂量的TMU4142即可完全逆转小鼠的抑郁样行为。相比之下,传统5-HT1AR激动剂gepirone和buspirone在相同实验中均未显示抗抑郁作用。新奇食物抑制实验(novelty-suppressed feeding, NSF)进一步表明,TMU4142在给药1小时后即可产生显著的抗抑郁效果;而经典抗抑郁药氟西汀在相同实验中需经两周给药方能起效。这些结果表明,TMU4142在动物模型中的起效时间要远快于目前流行的抗抑郁药物氟西汀,展现出快速且显著的抗抑郁作用。
最后,研究团队利用光纤记录技术实时监测了小鼠中缝背核(DRN)区域的5-HT浓度及5-HT能神经元活性变化。结果显示,在产生抗抑郁效果的剂量下(1.5 mg/kg),TMU4142并不会引起DRN中5-HT浓度下降;相反,传统5-HT1AR激动剂F-13714和buspirone则可迅速降低该区域5-HT水平。神经元活性监测结果与5-HT浓度变化趋势一致,说明TMU4142在治疗剂量下并不会激活DRN中5-HT1A自受体的负反馈调节机制。
综上,本研究通过系统的结构药理学分析,基于结构信息成功开发出一种具有GoA信号通路选择性且Gi3活性较弱的5-HT1AR激动剂。该类化合物展现出显著区别于现有5-HT1AR激动剂的独特药理学特征,能够选择性激活5-HT1A异源受体,同时避免中缝背核5-HT1A自受体的反馈抑制,为抑郁症治疗提供了一种具有广阔应用前景的新型药物开发策略。
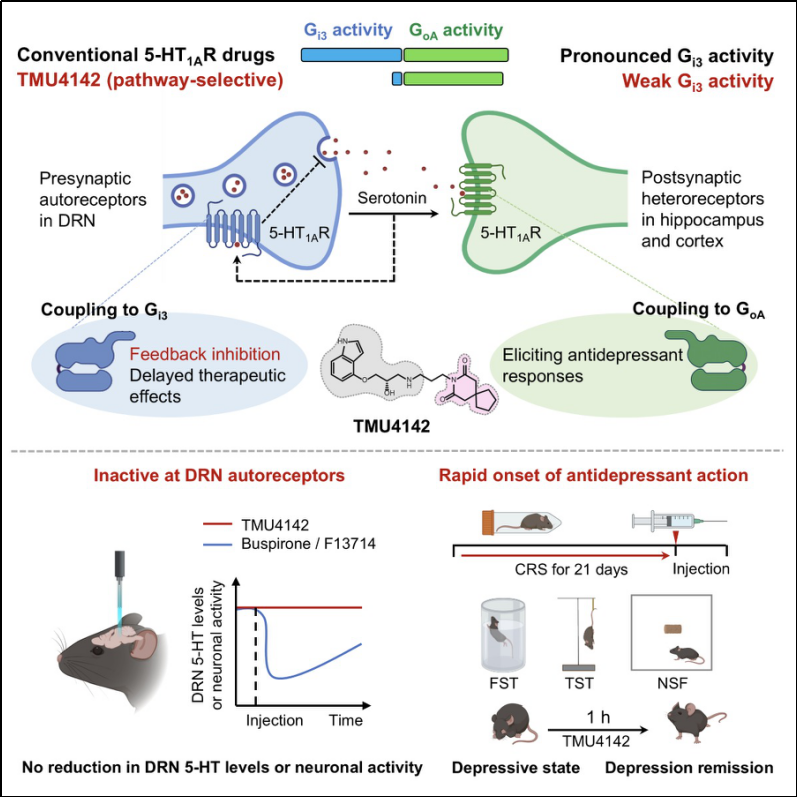
图1、基于GoA信号通路选择性5-HT1A受体部分激动剂的快速抗抑郁策略。
合肥综合性国家科学中心大健康研究院、免疫应答与免疫治疗全国重点实验室副研究员王春玉为该论文的第一作者兼通讯作者、中国科学技术大学生命科学与医学部博士研究生张楠、邵雨洁以及天津医科大学研究生李涛为本文的共同第一作者。合肥综合性国家科学中心大健康研究院、免疫应答与免疫治疗全国重点实验室、中国科学技术大学生命科学与医学部曹灿教授为最后通讯作者,中国科学技术大学生命科学与医学部薛天教授、史逸铭副研究员以及天津医科大学陈贺教授为共同通讯作者。团队成员张梦娜、高梦、王雨萌和梁亚琪等也为该项研究做出了重要的贡献。中国科学技术大学冷冻电镜中心高永翔博士为冷冻电镜数据采集提供了支持和帮助。
原文链接:https://doi.org/10.1016/j.cell.2025.10.022